2026
“Proximal Brønsted Acid Sites in Zeolites Cooperate to Stabilize Transition States via Hydrogen Bonding and Multi-Ion-Pair Interactions”
Submitted.

Abstract
Brønsted acid sites, when separated across molecular length scales, can cooperate via hydrogen-bonding and multi-ion-pair interactions to stabilize reactive intermediates and transition states during catalytic turnovers. Here, we study mechanisms underlying such inter-site cooperation using high-symmetry chabazite (CHA) framework zeolite catalysts, which provide a model platform to precisely synthesize and quantify acid sites in spatially discrete and proximal site arrangements. At low temperatures (<450 K), methanol dehydration to dimethyl ether (DME) occurs on H+ sites covered by methanol (0.2–52 kPa CH3OH), with experimentally measured turnover rates (per H+, 415 K) that are >8× higher, and activation enthalpies that are 25 kJ mol-1 lower, on proximal than isolated H+ sites. Density functional theory (DFT) calculations on H+ sites covered by adsorbed methanol provide evidence that proximal H+ sites facilitate hydrogen-bonding interactions via co-adsorbates that enthalpically stabilize the anionic conjugate bases of carbocations in dehydration transition states. Similarly, measured turnover rates (per H+, 748 K) are >7× higher during high-temperature (>700 K) protolytic alkane cracking and dehydrogenation on proximal than isolated H+ sites and DFT-optimized structures show evidence of hydrogen-bonding stabilization of carbocationic transition states at proximal acid sites. However, experimentally measured activation entropies (by ~25 J mol-1 K-1) and activation enthalpies (by ~10 kJ mol-1) are higher at proximal than isolated H+ sites, suggesting that proximal sites entropically stabilize (and enthalpically destabilize) transition states, the opposite behavior from DFT predictions from static (0 K) potential energy minimizations. Molecular dynamics (MD) simulations indicate that greater entropic freedom is conferred to guest cations positioned nearby proximal, rather than isolated, H+ sites due to multi-ion-pair interactions with conjugate lattice anions. These findings provide new mechanistic insights into how proximal acid sites cooperate via hydrogen-bonding interactions to enthalpically stabilize ion-pair transition states at low temperatures and via multi-ion-pair electrostatic interactions to entropically stabilize ion-pair transition states at high temperatures.
“Controlled Placement of Trivalent Heteroatoms in MFI Zeolite Frameworks Using Structure-Directing Agents”
Submitted.

Abstract
MFI zeolites contain distinct confining pore environments, either smaller (~0.55 nm) channels or larger (~0.70 nm) intersections. The substitution of trivalent heteroatoms generates Brønsted acid sites of varying acid strength, which together with their location within different pore environments, dictates the stability of transition states via electrostatic (ion-pair) and van der Waals interactions. Here, we report that structure-directing agents (SDAs) that control Al3+ substitutional patterns during MFI crystallization analogously position other trivalent heteroatoms (Ga3+, Fe3+, B3+), altering the distribution of acid sites between channels and intersections, as probed by low-temperature (403 K) toluene methylation kinetics. Heteroatom-substituted MFI crystallized with tetrapropylammonium result in low selectivity towards p-xylene (<30%), while those crystallized using (co-)SDAs containing peripheral hydrogen-bonding groups (e.g., ethylenediamine and tetrapropylammonium) show high p-xylene selectivity (~80%). These selectivities are consistent with DFT-calculated Gibbs free energy barriers for intersection-dominant and channel-dominant active site distributions, respectively. Transition states to form each xylene isomer are similar in size and charge; thus, their rate constants decrease with acid strength similarly, causing isomer selectivity to depend strongly on confinement but not on acid strength. These generalizable synthetic strategies enable independently controlling acid site strength and location in MFI zeolites and, in turn, catalytic rates and selectivities.
“High-yield oxidative propylene production via chemical looping”
Submitted.

Abstract
The oxidative conversion of propane to propylene presents a promising route to energy-efficient olefin production, but current catalysts fail to attain yields above 30% without concurrent direct dehydrogenation, which diminishes the value of proposition. Here, we report a one-step, isothermal process that achieves ~60% propylene yield with near-quantitative hydrogen removal by coupling a propane dehydrogenation (PDH) catalyst with a selective hydrogen combustion (SHC) material within a chemical looping (CL) framework. The PDH catalyst consists of quasi-atomically dispersed bimetallic PtSn clusters embedded in a K-MFI zeolite host that maintain high activity and selectivity over multiple reaction cycles, without requiring conventional hydrogen pre-activation—a critical feature when integrated with the SHC component. The SHC material (LaMnO3 perovskite) exhibits high stability, large oxygen storage capacity, rapid hydrogen combustion kinetics, and strong selectivity against hydrocarbon oxidation. Together, the tandem CL-DH+SHC system achieves ~78% propane conversion, ~81% propylene selectivity, and ~90% hydrogen removal efficiency—tripling the yield achieved by state-of-the-art ODH catalysts. This strategy overcomes a long-standing tradeoff between propylene yield and hydrogen conversion by combining optimized DH and SHC components within a CL configuration that eliminates the need for energy-intensive air separation units, advancing, thus, a viable route to single-pass, high-yield oxidative olefin production.

“Predicting Hydrogen Saturation on Small Transition Metal Nanoparticles”
Journal of Physical Chemistry C, 130 (2026), 4398–4412.

Abstract
Hydrogen adsorption is important for understanding coverage effects for various chemical reactions and for characterizing the size of nanoparticles. Recent literature suggests that the often-assumed saturation coverage of unity may not be reasonable, at least for small nanoparticles. This includes our prior study which showed that Ir and Pt nanoparticles, which primarily bind H adsorbates in atop binding modes, can saturate at H/Msurf ratios well above 1, depending on the metal and particle size. This work examines H adsorption on small nanoparticles of Ru, Rh, Pd, and Os metals that preferentially bind H in 3-fold sites. We therefore investigate new adlayer structures to understand how undercoordinated atoms influence these H configurations. Calculations on 201-atom nanoparticles suggest H* (at low coverages) binds most favorably to undercoordinated bridge sites for all six metals of interest. As suggested by the slab model calculations, 3-fold sites are also among the most stable sites for Ru, Rh, and Pd particles, while atop sites are more stable for Os, Ir and Pt. These trends in single H* binding behavior do not significantly change with particle size. Saturation adlayers for 4d metals (Ru, Rh, and Pd) are dominated by H* bound to fcc 3-fold sites and undercoordinated bridge sites. With saturation coverages of 1.38, 1.48, and 1.28 ML respectively, Rh has the highest saturation coverage of the 4d metals, reflecting a non-monotonic periodic trend. For the 5d metals (Os, Ir, Pt), saturated adlayers are dominated by the same undercoordinated bridge sites, but also generally favor atop-bound H* instead of 3-fold except Pt. Their saturation coverages are 1.87, 1.93, and 1.57 ML, respectively. Saturation coverage increases when moving from a 4d to a 5d metal, and the Group 9 metals (Rh and Ir) exhibit the highest saturation in their respective series. Our results show that H* binding energy is largely independent of particle size for a given site type. Instead, changing the particle size primarily alters the distribution of available binding sites (corner, edge, and terrace). Therefore, the adlayer model developed for the 201-atom particle can be used to reliably estimate saturation coverages, allowing for extrapolation to both smaller and larger nanoparticles.

“Tunable surface cation arrangements in IrxRu1−xO2(110) and their impact on adsorbate binding”
ACS Catalysis, 16 (2026) 5038–5053

Abstract
Advancing methods to control and characterize the local cation arrangements on mixed metal oxide surfaces can afford opportunities for designing catalysts with tunable chemical properties. Here, we show that IrxRu1−xO2(110) solid solutions with variable near-surface cation arrangements can be synthesized by annealing IrO2/RuO2 layered structures under mild oxidizing conditions in ultrahigh vacuum. Temperature-programmed desorption (TPD) of adsorbed N2 and atomic O provides a quantitative probe of the Ir and Ru populations in cus binding sites (Mcus) and their nearest-neighbor subsurface sites (Msubcus), due to the strong, highly localized influence of these cations on N2 and O binding. The resulting Mcus/Msubcus configurations remain stable up to at least 650 K, the temperature range relevant for catalytic studies. Heating IrO2/RuO2 layered structures to ~800 K induces rapid Ru–Ir intermixing and drives Ru enrichment in the near-surface region. TPD measurements show that Ru strongly favors incorporation into cus-sites during our heating protocol and begins to occupy subcus-sites only after the Rucus population approaches saturation. After extended heating, the surface is dominated by Rucus over Rusubcus motifs (Ru/Ru2) characteristic of pure RuO2(110), with small populations of Ru/Ir2 and Ru/IrRu. Density functional theory (DFT) predicts binding-site distributions for O-terminated surfaces that closely match those determined from TPD. DFT reveals that enhanced O binding on Rucus relative to Ircus, along with stabilization by neighboring Irsubcus atoms, drives Ir–Ru interchange to selectively form Ru/Ir2 motifs and promote Ru occupation of subcus sites after cus sites are filled. Together, these results provide new insight into the synthesis and characterization of mixed IrxRu1−xO2(110) surfaces and demonstrate that controlling near-surface cation arrangements offers a promising route to tuning the chemical properties of mixed-oxide catalysts.

“Polymethylated aromatics deactivate intracrystalline acid sites in MFI zeolites during low-temperature toluene methylation”
Journal of Catalysis, 454 (2026) 116653.

Abstract
Toluene methylation on Brønsted acidic aluminosilicates at low temperatures (<433 K) and conversions (<1%) show initial rates and selectivities dictated by the stabilities of transition states confined within voids of molecular dimensions, but deactivation leads to decreases in rates and shifts in selectivity according to mechanisms that remain incompletely understood. Here, we elucidate the predominant mechanism by which MFI zeolites deactivate and provide insights into the identity and location of deactivating species. Gas-phase aromatic product distributions formed on aluminosilicates of varying pore size (MFI, BEA, FAU, MCM-41) during toluene methylation (403 K, 4 kPa toluene, 66 kPa dimethyl ether) indicate that methylbenzenes larger than xylenes and 1,2,4-trimethylbenzene form, but are unable to egress from medium-pore (~0.55 nm diameter) MFI zeolites. Co-feeding a selective poison of extracrystalline H+ sites (e.g., 2,6-di-tert-butylpyridine) reveals that intracrystalline H+ sites in MFI zeolites dominate observed deactivation behavior; as a result, deactivation-induced changes to xylene selectivity reflect disproportionate decreases in the numbers of intracrystalline H+ sites that remain active, relative to extracrystalline H+ sites. Post-reaction thermal treatments (up to 853 K) of deactivated MFI zeolites result in the concomitant formation of C6–10 monocyclic aromatic hydrocarbons and C2–3 aliphatic hydrocarbons, and are able to fully regenerate MFI zeolites. These results implicate bulky polymethylbenzenes (on average, C11) as predominant deactivating species that cannot egress from crystallites, and only dealkylate at higher temperatures. These findings provide clarity on how zeolites deactivate and engender changes in selectivity during low-temperature toluene methylation, and provide guidance for zeolite catalyst design and regeneration.
“Nanoparticle size and composition of PdAu catalysts govern the structure and kinetics of H2O2 formation through O2 reduction”
Journal of Catalysis, 454 (2026) 116649.

Abstract
The identities and distribution of active site motifs on bimetallic nanoparticle surfaces depend upon their composition and size, and these factors impact rates and selectivities of surface catalyzed reactions. We combine steady-state rate measurements of O2-H2 reactions to form H2O2 and H2O, ex situ spectroscopy, and density functional theory (DFT) calculations to elucidate the structure-performance relationships of silica-supported PdχAu catalysts (1.8 and 11.8 nm diameters, where χ describes the Pd mole fraction). Smaller (1.8 nm) PdχAu nanoparticles display greater H2O2 selectivities and rates relative to larger (11.8 nm) particles regardless of bulk composition, which suggests the number and coordination of exposed Pd atoms dictates these trends. Ex situ infrared spectra of adsorbed CO (CO*) and mixed 12CO*–13CO* adlayers combined with DFT-calculated structures and vibrational frequencies reveal greater Pd isolation on smaller nanoparticles. This is evidenced by weaker dipole coupling interactions between CO* species, likely due to differences in particle curvature and greater fractions of surface sites with stable binding upon nanoparticles of smaller diameters (but equivalent bulk composition). Molecular interpretations of measured apparent activation enthalpies further confirm Pd active site structure differences with nanoparticle size given the same Pd content, reflecting changes in H2O2 selectivities. H2O2 selectivities remain largely constant with reactant pressures on 11.8 nm PdχAu catalysts but vary significantly with H2 to O2 pressure ratios on 1.8 nm PdχAu catalysts. These differences show the coordination of Pd atoms in smaller nanoparticles respond more sensitively to reactant coverages, and high O2 chemical potentials stabilize Pd atoms at the surfaces. Collectively, these results demonstrate how nanoparticle size affects the location, size, and spatial distributions of Pd ensembles, and nanoparticle surface restructuring in response to changes in reaction conditions—ultimately governing intrinsic kinetics and selectivity to H2O2.
2025
“Hydrothermally Stable and Regenerable Molybdenum-Zeolite Catalysts for Non-oxidative Conversion of Methane to Dihydrogen and Ethene”
ACS Catalysis, 15 (2025) 17568–17580.
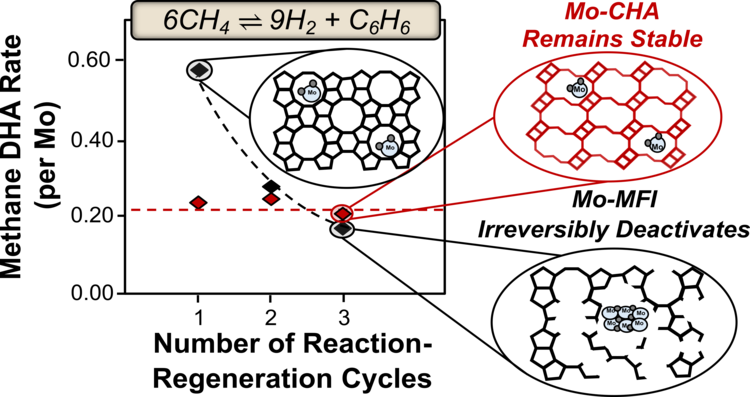
Abstract
Designing Mo-zeolite catalysts that retain structural stability over many reaction-regeneration cycles is a challenge in developing practical methane dehydroaromatization (DHA) routes to produce dihydrogen, ethene, and aromatics. Mo-MFI zeolites typically used for methane DHA irreversibly deactivate during regeneration due to framework dealumination and structural degradation. Here, we synthesize Mo supported on small-pore zeolites (e.g., CHA, AEI, RTH), which are more durable than MFI under hydrothermal aging conditions (823 K, >2 kPa H2O). Methane DHA forms H2 and ethene at equivalent rates on Mo-CHA and Mo-MFI. Aromatics formation rates (per Mo) are intracrystalline-diffusion limited in CHA, but increase with decreasing CHA crystallite size. Initial DHA rates (per Mo) remain invariant on Mo-CHA after successive reaction-regeneration cycles (>10), supported by high-resolution TEM and quantitative site characterization data showing minimal site or structural degradation on Mo-CHA, in sharp contrast to the systematic decrease in rates and structural degradation observed on Mo-MFI. This work reports materials that can be fully regenerable under potential industrial conditions and provides structure-function relations between catalysts properties and DHA rates, selectivity, and long-term stability.
“Pentaerythritol Structure-Directing Agents Bias Aluminum Siting in MFI Zeolites to Increase Para-Xylene Selectivity during Toluene Methylation”
Journal of Catalysis, 450 (2025) 116470.
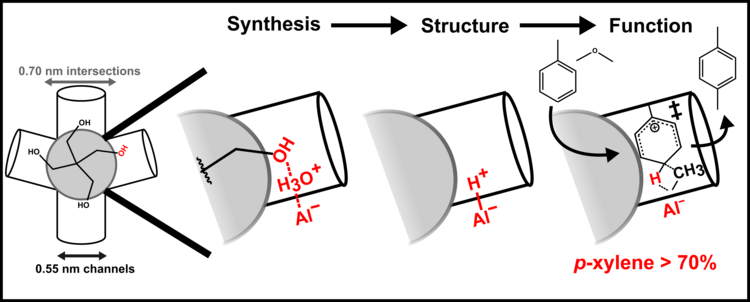
Abstract
“DFT+U Calculations on Substitutionally Doped (Ni, Cu, Zn) Mg-vanadate Surfaces for the Oxidative Dehydrogenation of Alkanes”
Journal of Catalysis, 450 (2025) 116313.
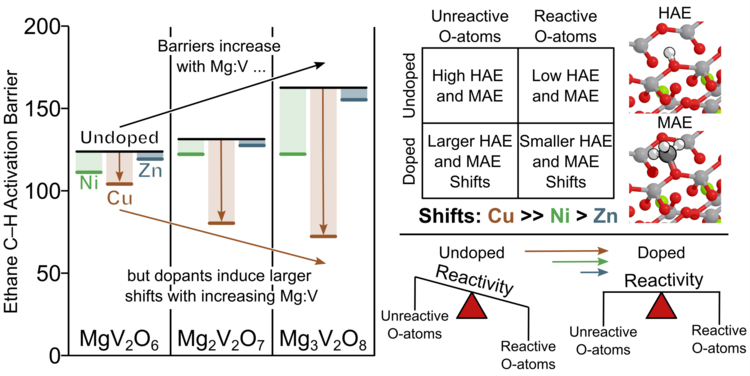
Abstract
“Modified adsorption energies on single-layer IrO2 and RuO2 films of IrO2-RuO2 heterostructures: Localized effect of subsurface metal-oxygen ligands”
ACS Catalysis, 15 (2025) 11134–11149.
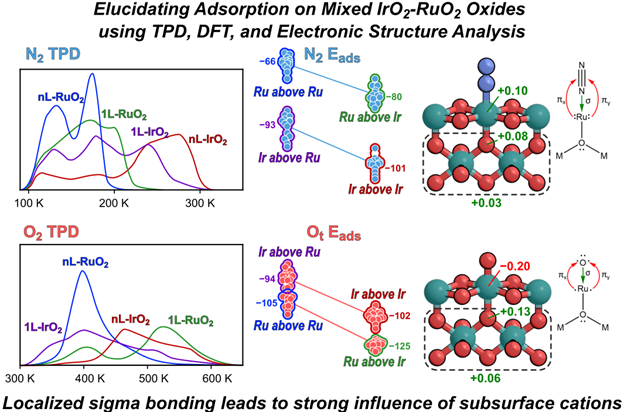
Abstract
“Predicting a Generalized Mechanism of Branched Hydrogenolysis on Ru, Ir, and Pt Surfaces for Polymer Upcycling Applications”
Journal of Catalysis, 450 (2025) 116200.
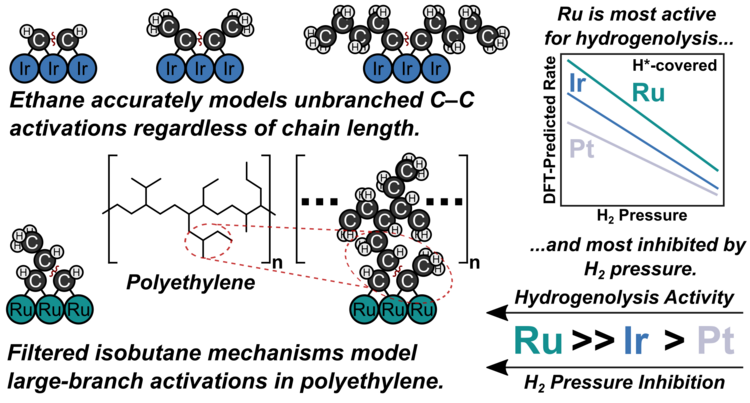
Abstract
“Assessing the Influence of Void Environment in MFI Zeolites on Propene Oligomerization Kinetics Using a Combined Computational and Experimental Approach”
ACS Catalysis, 15 (2025) 7121–7137.
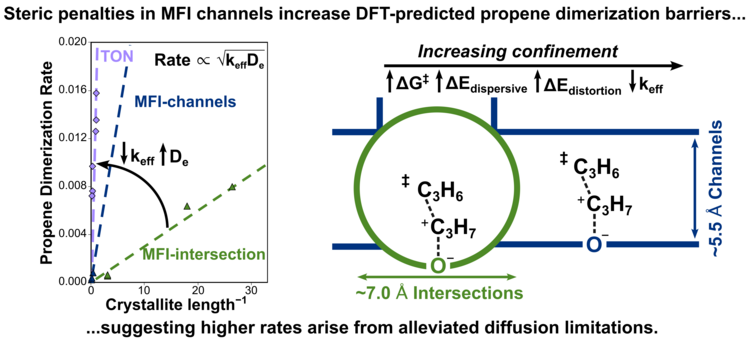
Abstract
“Understanding AuPd Alloy Nanoparticle Structure under Vacuum Using DFT and Monte Carlo Methods”
Journal of Physical Chemistry C, 129 (2025), 5702–5717.
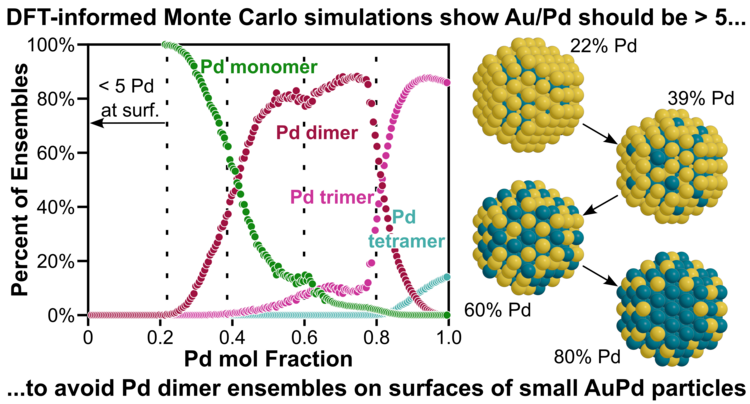
Abstract
2024
“A Career in Catalysis: Enrique Iglesia”
ACS Catalysis, 14 (2024) 10583–10613.
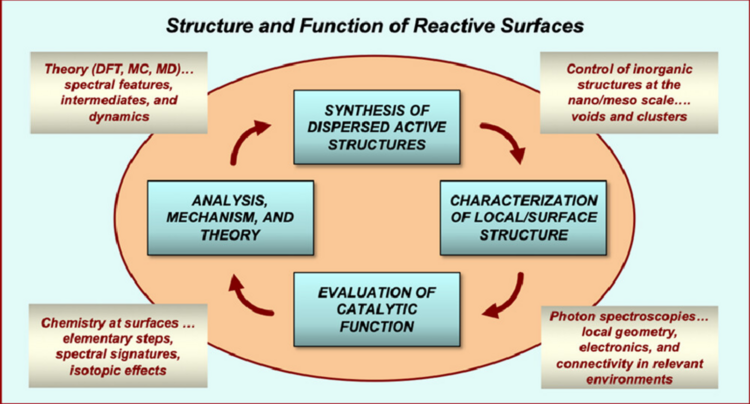
Abstract

“Synthetic Placement of Active Sites in MFI Zeolites for Selective Toluene Methylation to para-Xylene”
Journal of the American Chemical Society, 146 (2024) 10666–10678.
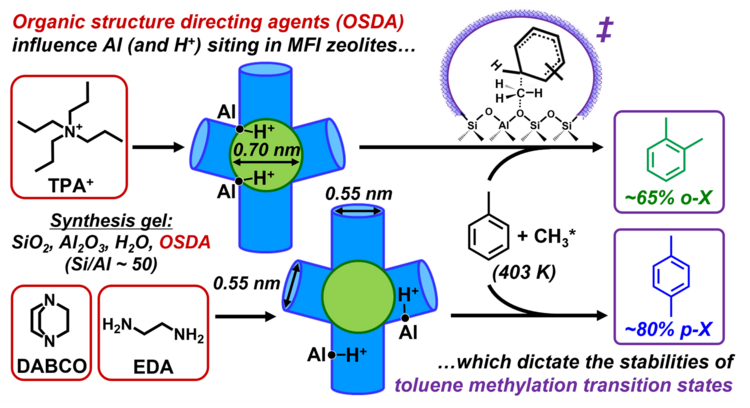
Abstract
“Predicting the Enthalpy of Hydrocarbon Radicals Adsorbed on Pt(111) Using Molecular Fingerprints and Machine Learning”
Journal of Physical Chemistry C, 128 (2024) 5030–5043.
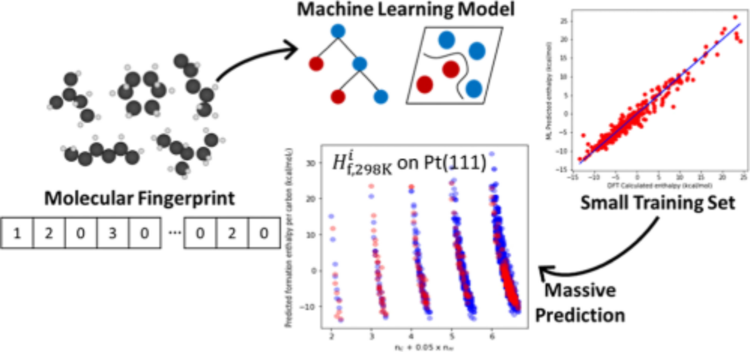
Abstract
“Kinetics and Reaction Mechanism of Pd-Catalyzed Chlorobenzene Hydrogenolysis”
Journal of Catalysis, 432 (2024) 115435.
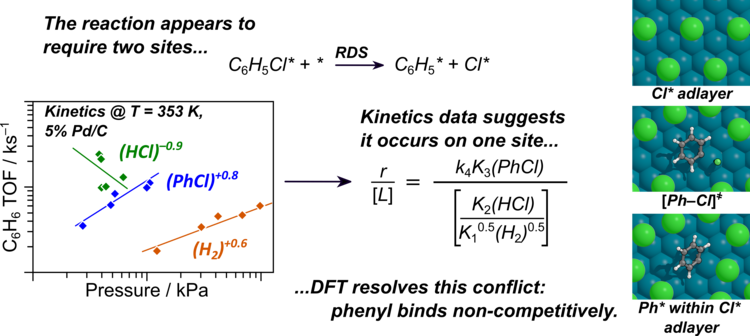
Abstract
“Electronic and Geometric Features Controlling the Reactivity of Mg-vanadate and V2O5 Surfaces toward the Initial C–H Activation of C1–C3 Alkanes – a DFT+U Study”
Journal of Catalysis, 442 (2025) 115800.
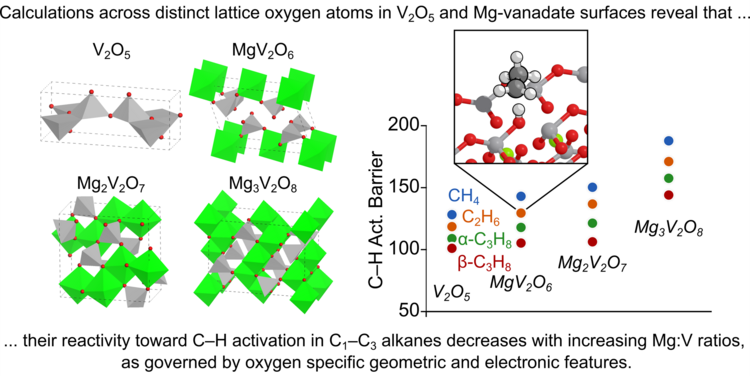
Abstract
2023
“Role of Phosphorous in Transition Metal Phosphides for Selective Hydrogenolysis of Hindered C–O Bonds”
Journal of Catalysis, 421 (2023) 403–418.
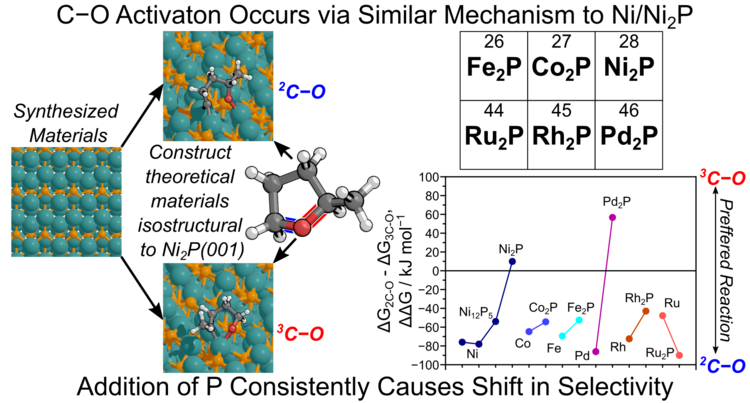
Abstract
“Brønsted Acid Strength Does Not Change for Bulk and External Sites of MFI”
ACS Catalysis, 13 (2023) 4470–4487.
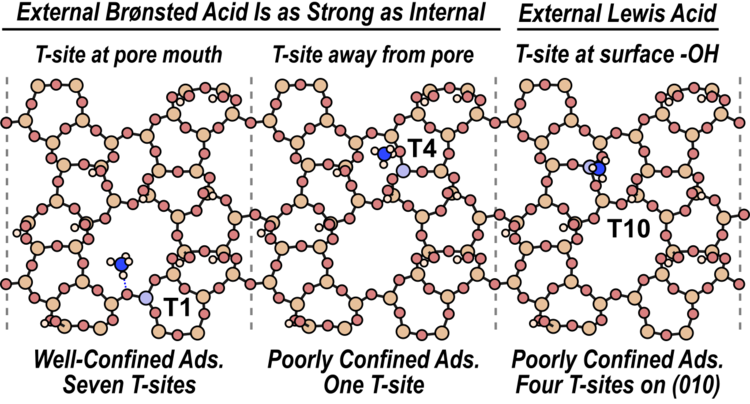
Abstract
“Mechanisms and Kinetics of the Dehydrogenation of C6-C8 Cycloalkanes, Cycloalkenes, and Cyclodienes to Aromatics in H-MFI Zeolite Framework”
ACS Catalysis, 13 (2023) 99–112.
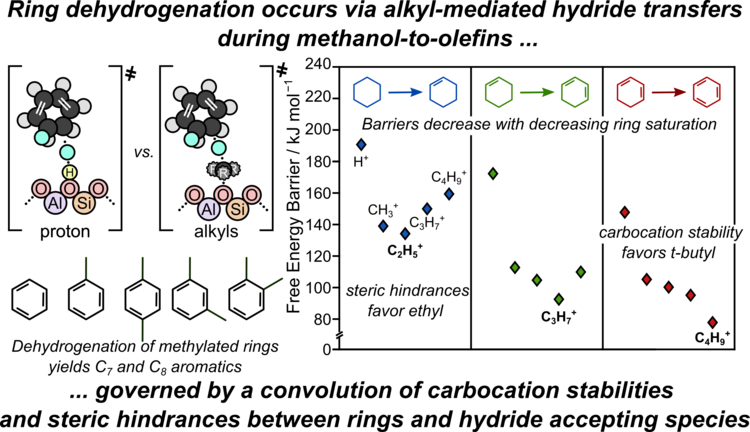
Abstract
2022
“Experimental and theoretical characterization of Rh single-atoms supported on γ-Al2O3 with varying hydroxyl content during NO reduction by CO”
ACS Catalysis, 12 (2022) 11697–11715.
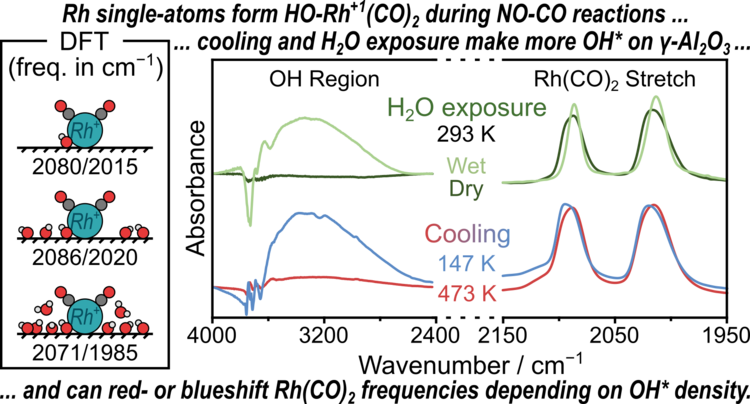
Abstract

“Altering the Arrangement of Framework Al Atoms in MEL Zeolites Using Mixtures of Tetrabutylammonium and Sodium Structure-Directing Agents”
Chemistry of Materials, 34 (2022) 6835–6852.
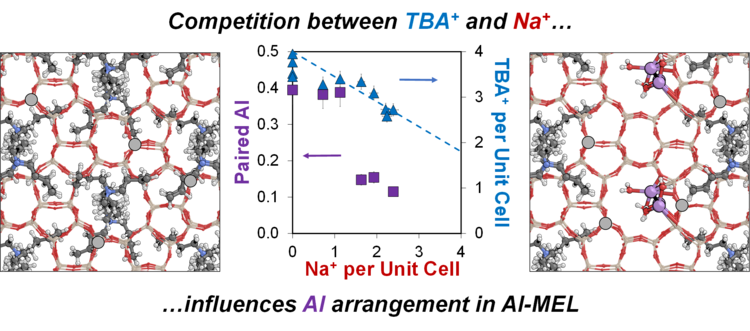
Abstract
“Oxygen-Doped Carbon Supports Modulate the Hydrogenation Activity of Palladium Nanoparticles through Electronic Metal−Support Interactions”
ACS Catalysis, 12 (2022) 7344–7356.

Abstract
“Binding and Exchange Reactions of Hydrogen Isotopes on Surfaces of Dispersed Pt Nanoparticles”
Journal of Physical Chemistry C, 126 (2022), 3923–3938.
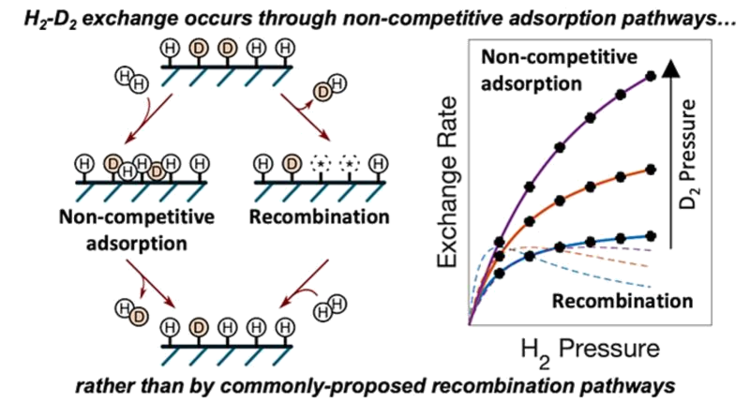
Abstract
“Predicting diffusion barriers and diffusivities of C6–C12 methylbenzenes in MFI zeolites”
Microporous and Mesoporous Materials, 333 (2022), 111705.
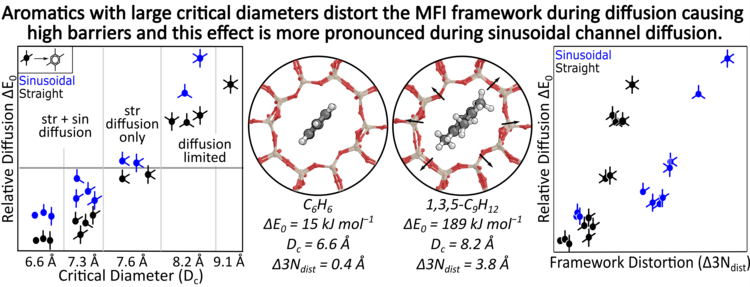
Abstract
“The Fischer-Tropsch synthesis: A few enduring mechanistic conundrums revisited”
Journal of Catalysis, 405 (2022) 614–625. Perspective
First Paragraph
2021

“Quantifying effects of active site proximity on rates of methanol dehydration to dimethyl ether over CHA zeolites through microkinetic modeling”
ACS Materials Au, 2 (2021), 163–175.
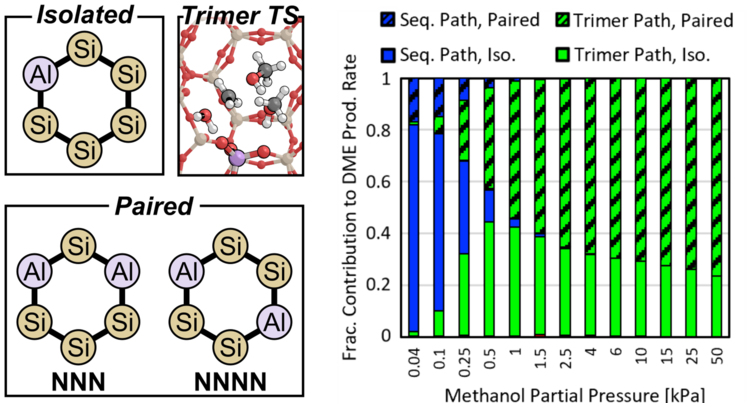
Abstract
“Theoretical and experimental characterization of adsorbed CO and NO on γ-Al2O3-supported Rh nanoparticles”
Journal of Physical Chemistry C, 125 (2021) 19733–19755.
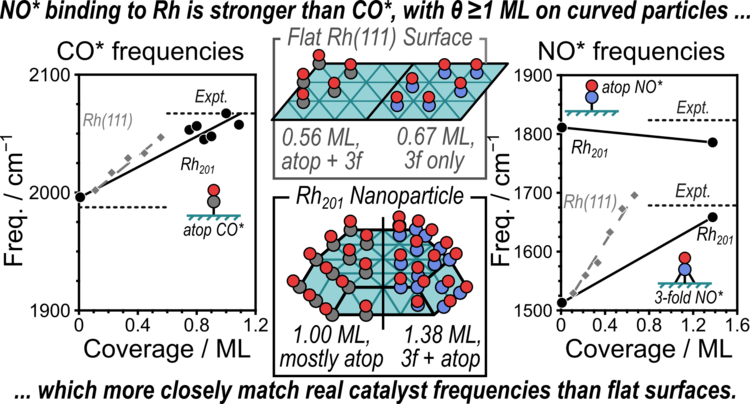
Abstract
“Comparing Alkene Disproportionation and Formaldehyde-mediated Diene Formation Routes in Methanol-to-Olefins Catalysis in MFI and CHA”
Journal of Catalysis, 400 (2021) 124–139.
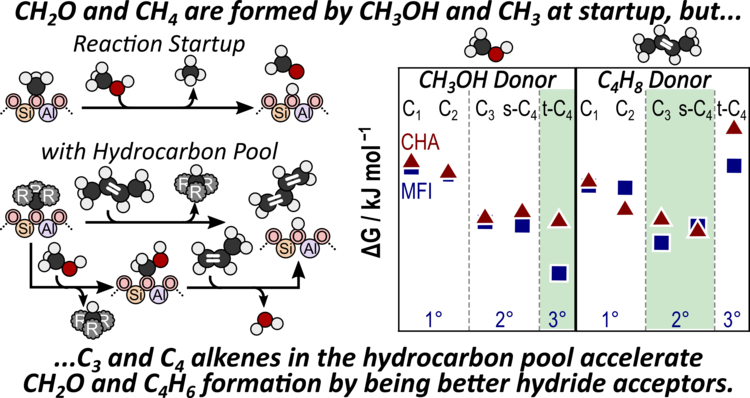
Abstract

“Hydrogenation and C-S bond activation pathways in thiophene and tetrahydrothiophene reactions on sulfur-passivated surfaces of Ru, Pt, and Re nanoparticles”
Applied Catalysis B: Environmental , 291 (2021) 119797.
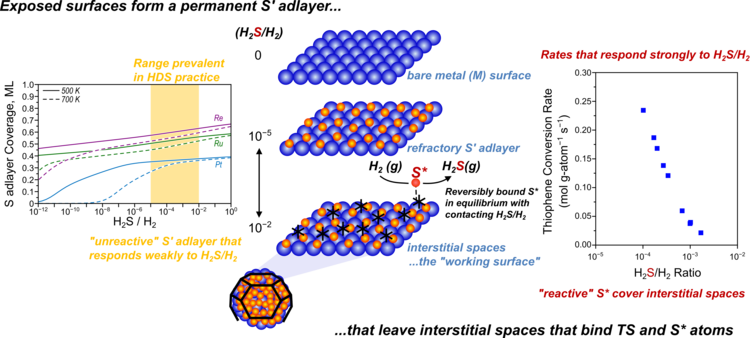
Abstract
2020
“Oxophilicity Drives Oxygen Transfer at a Palladium-Silver Interface for Increased CO Oxidation Activity”
ACS Catalysis, 10 (2020) 13878–13889.
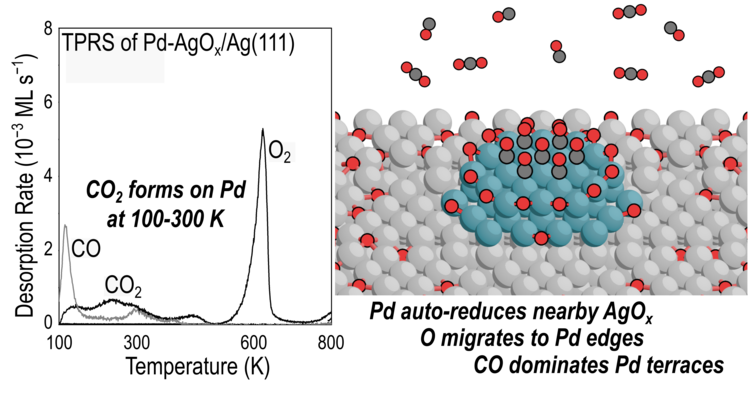
Abstract
“Experimental and Theoretical Assessments of Aluminum Proximity in MFI Zeolites and its Alteration by Organic and Inorganic Structure-Directing Agents”
Chemistry of Materials, 32 (2020) 9277–9298.
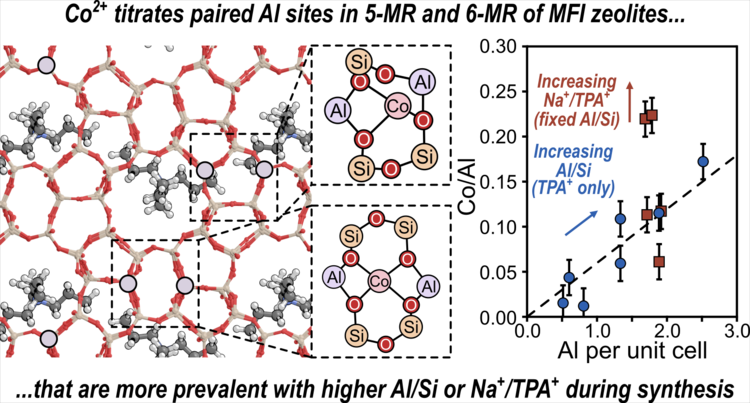
Abstract

“Rigid Arrangements of Ionic Charge in Zeolite Frameworks Conferred by Specific Al Distributions Preferentially Stabilize Alkanol Dehydration Transition States”
Angewandte Chemie Int. Ed., 59 (2020) 18686–18694.
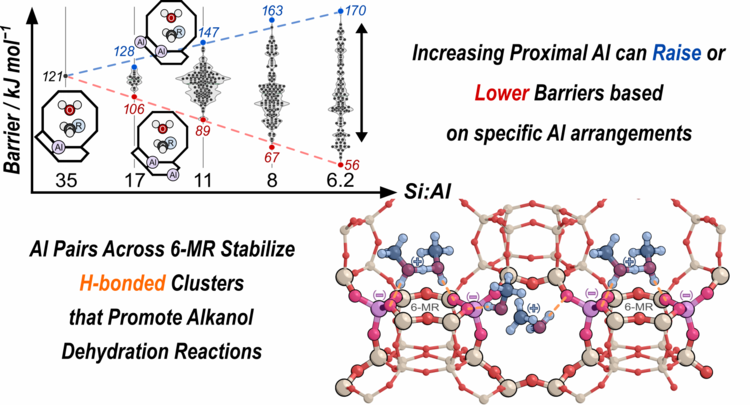
Abstract
“Highly Selective Cross-Etherification of 5-Hydroxymethylfurfural with Ethanol”
ACS Catalysis, 10 (2020) 6771–6785.
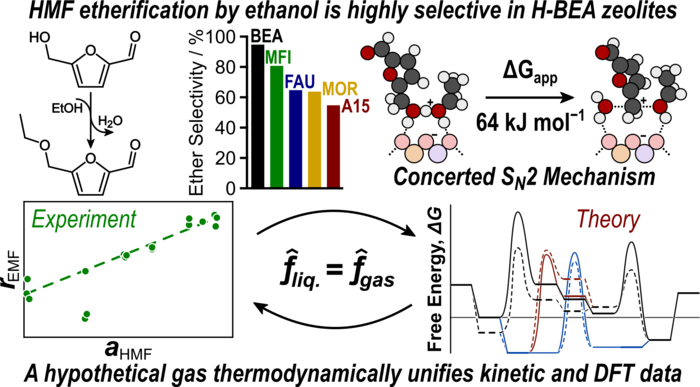
Abstract
“Mechanism and Effects of Coverage and Particle Morphology on Rh and Pt Catalyzed NO-H2 Reactions”
Journal of Physical Chemistry C, 124 (2020) 13291–13303.
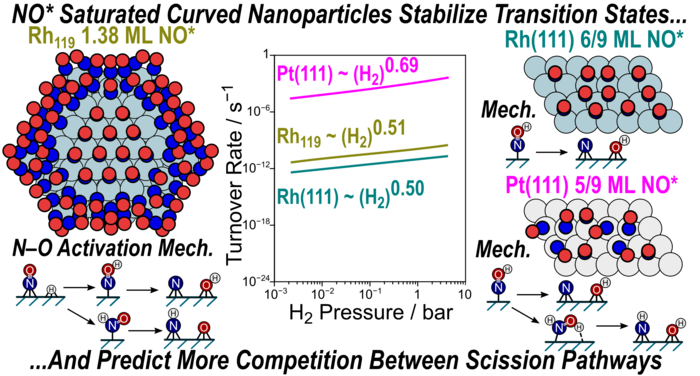
Abstract
“Contrasting Arene, Alkene, Diene, and Formaldehyde Hydrogenation in H-ZSM-5, H-SSZ-13, and H-SAPO-34 Zeolite Frameworks during MTO”
ACS Catalysis, 10 (2020) 4593–4607.
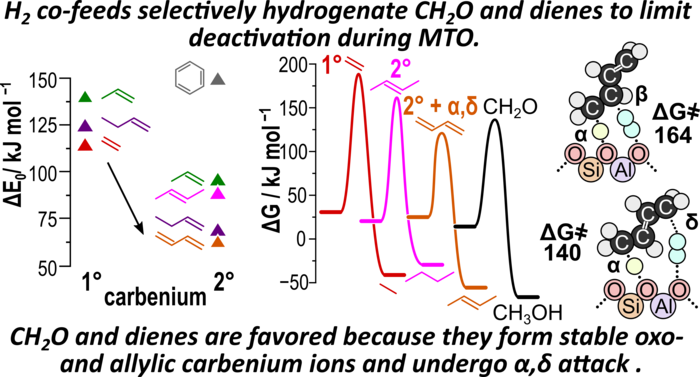
Abstract
32. A. Almithn and D. Hibbitts*
“Impact of Metal and Heteroatom Identities in the Hydrogenolysis of C–X Bonds (X = C, N, O, S, and Cl)”
ACS Catalysis, 10 (2020) 5086–5100.
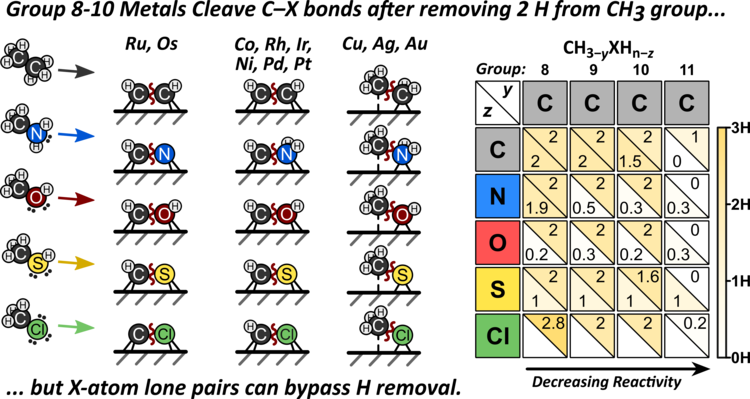
Abstract
2019
31. J. Di Iorio, A. Hoffman, C. Nimlos, S. Nystrom, D. Hibbitts*, and R. Gounder*
“Mechanistic Origins of the High-Pressure Inhibition of Methanol Dehydration Rates in Small-Pore Acidic Zeolites.”
Journal of Catalysis, 380 (2019) 161–177.
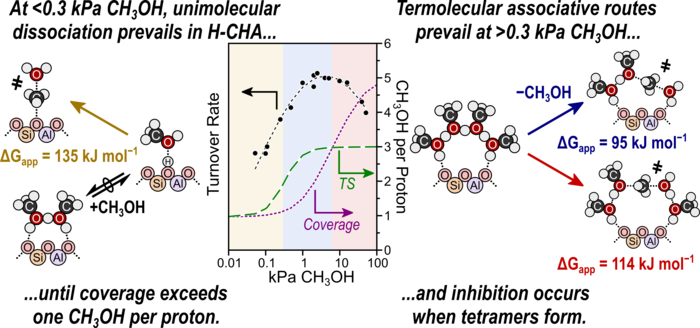
Abstract

30. P. Kravchenko, C. Plaisance, and D. Hibbitts*,
“A New Computational Interface for Catalysis.”
Pre-print, (2019).
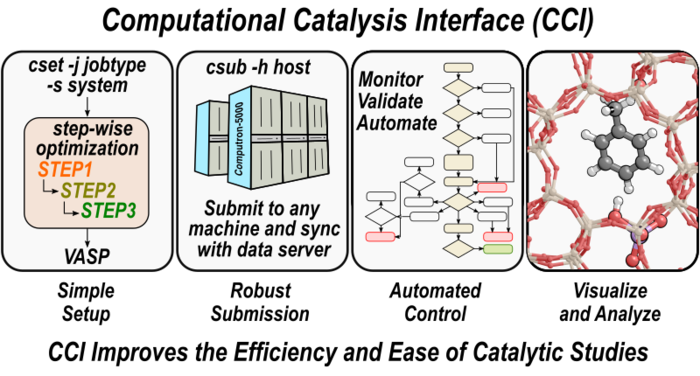
Abstract
29. M. DeLuca and D. Hibbitts*,
“Prediction of C6–C12 Interconversion Rates Using Novel Zeolite-specific Kinetic Monte Carlo Simulation Methods.”
Pre-print, (2019).
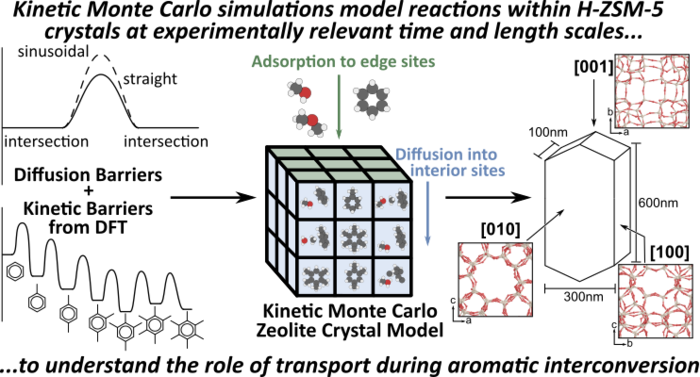
Abstract
28. M. Witzke, A. Almithn, C. Coonrod, M. Triezenberg, D. Hibbitts*, and D. Flaherty*,
“In situ Spectroscopic Methods for Isolating Reactive Intermediate Structures during Hydrogenolysis Reactions.”
Journal of the American Chemical Society, 141 (2019) 16671–16684.
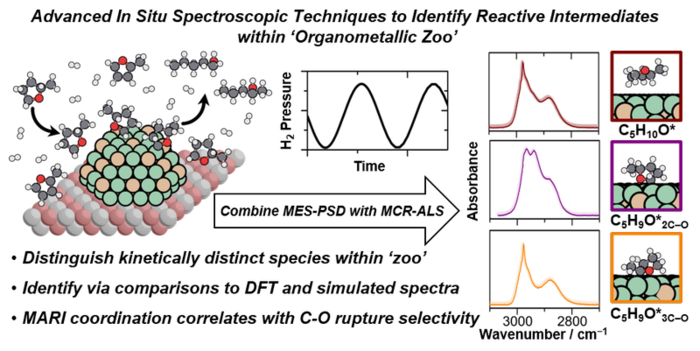
Abstract
27. M. DeLuca, P. Kravchenko, A. Hoffman, and D. Hibbitts*,
“Mechanism and Kinetics of Methylating C6–C12 Methylbenzenes with Methanol and DME in H-MFI Zeolites.”
ACS Catalysis, 9 (2019) 6444–6460.
Front Cover Article!
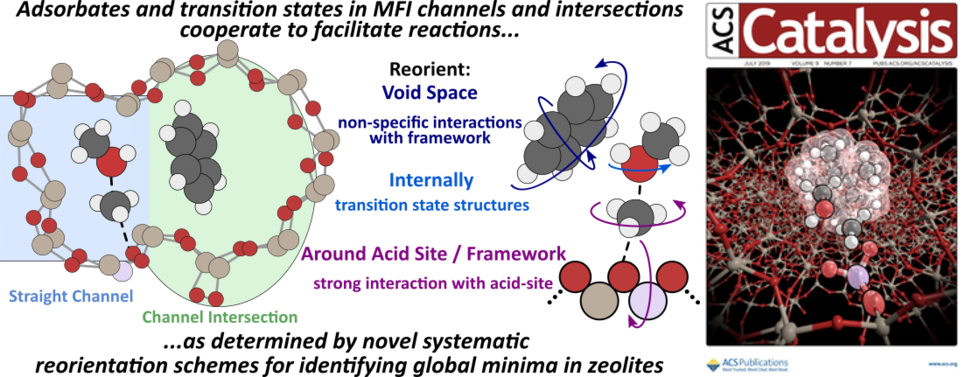
Abstract
26. A. Hoffman, M. DeLuca, and D. Hibbitts*,
“Restructuring of MFI Framework Zeolite Models and their Associated Artifacts in Density Functional Theory Calculations.”
Journal of Physical Chemistry C, 123 (2019) 6572–6585.
Editor's Choice!
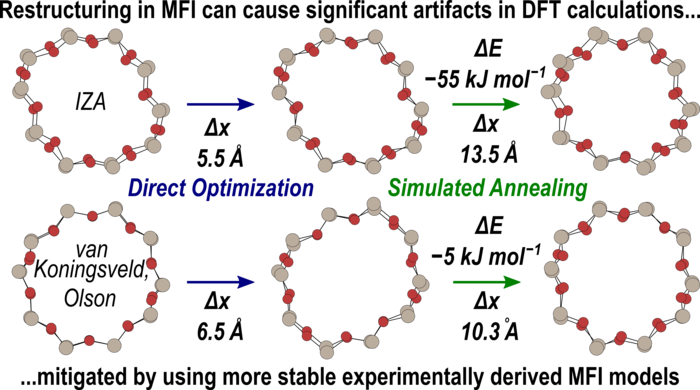
Abstract
25. A. Almithn and D. Hibbitts*,
“Comparing Rate and Mechanism of Ethane Hydrogenolysis on Transition Metal Catalysts.”
Journal of Physical Chemistry C, 123 (2019) 5421–5432.
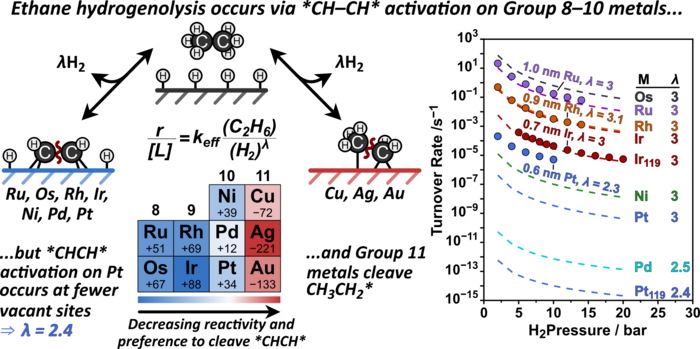
Abstract
24. M. Garcia-Dieguez, D. Hibbitts*, and E. Iglesia*,
“Hydrogen Chemisorption Isotherms on Pt Particles at Catalytic Temperatures: Langmuir and Two-Dimensional Gas Models Revisited.”
Journal of Physical Chemistry C, 123, (2019), 8447–8462.
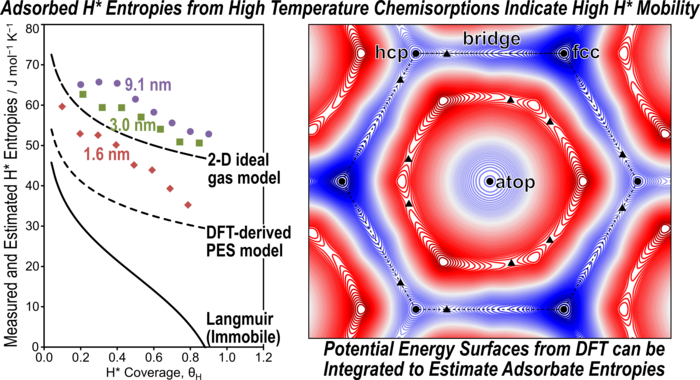
Abstract
2018
23. M. Cordon, J. Harris, J. Vega-Vila, J. Bates, S. Kaur, M. Gupta, M. Witzke, E. Wegener, J. Miller, D. Flaherty, D. Hibbitts, R. Gounder*,
“The Dominant Role of Entropy in Stabilizing Sugar Isomerization Transition States within Hydrophobic Zeolite Pores.”
Journal of the American Chemical Society, 140 (2018) 14244–14266.
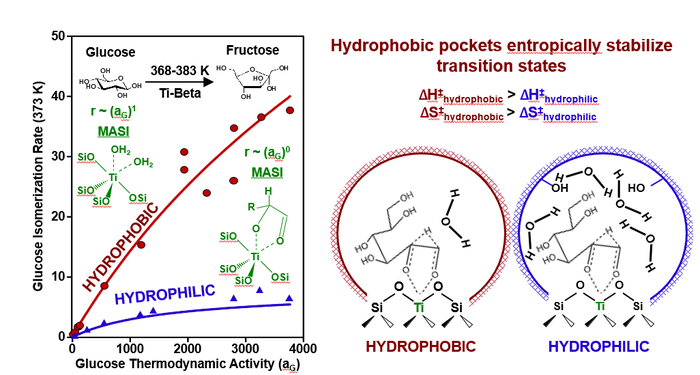
Abstract
22. S. Nystrom^, A. Hoffman^, and D. Hibbitts*,
“Tuning Brønsted Acid Strength by Altering Site Proximity in CHA Framework Zeolites.”
ACS Catalysis, 8 (2018) 7842–7860.
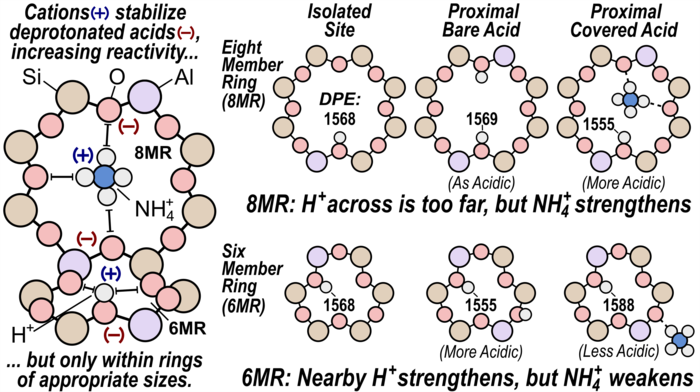
Abstract
21. M. Witzke, A. Almithn, C. Coonrod, D. Hibbitts*, and D. Flaherty*,
“Mechanisms and Active Sites for C-O Bond Rupture within 2-Methyltetrahydrofuran over Nickel Phosphide Catalysts.”
ACS Catalysis, 8 (2018) 7141–7157.
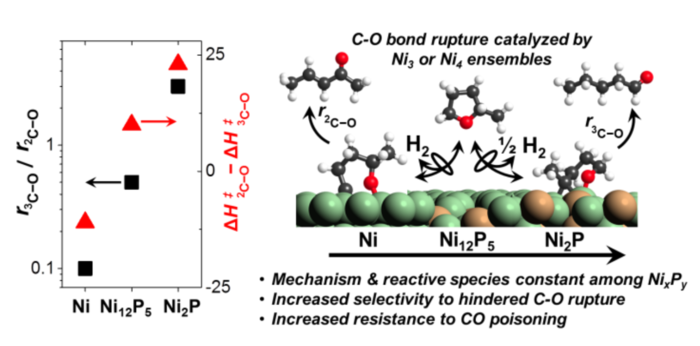
Abstract
20. A. Almithn and D. Hibbitts*,
“Effects of Catalyst Model and High Adsorbate Coverages in ab initio Studies of Alkane Hydrogenolysis.”
ACS Catalysis, 8 (2018) 6375–6387.
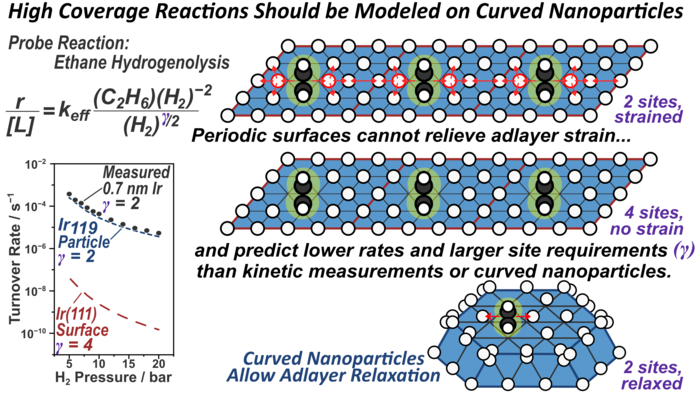
Abstract

19. A. Almithn and D. Hibbitts*,
“Supra-Monolayer Coverages on Small Metal Clusters and Their Effects on H2 Chemisorption Particle Size Estimates.”
AIChE Journal, 64 (2018) 3109–3120. Invited.
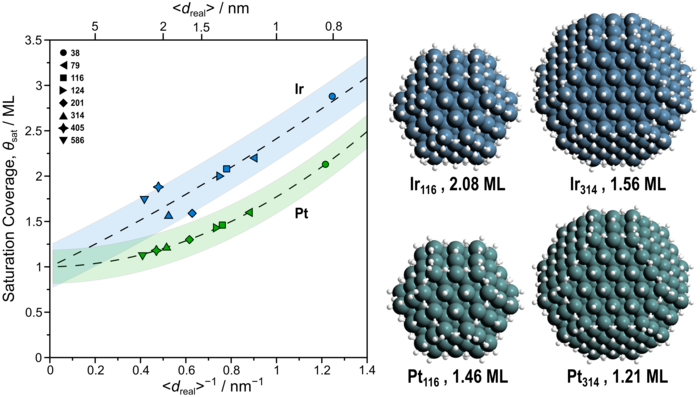
Abstract
2017
18. J. Liu, D. Hibbitts*, and E. Iglesia*,
“Dense CO Adlayers as Enablers of CO Hydrogenation Turnovers on Ru Surfaces.”
Journal of the American Chemical Society, 139 (2017) 11789–11802.
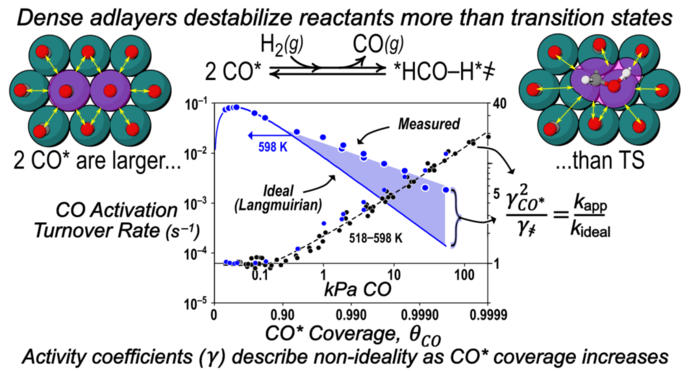
Abstract

17. R. Rao, R. Blume, T, Hansen, E. Fuentes, K. Dreyer, S. Moldovan, O. Ersen, D. Hibbitts, Y. Chabal, R. Schlogl, and J. Tessonnier*,
“Interfacial charge distributions in carbon-supported palladium catalysts.”
Nature Communications, 8 (2017) 340:1–10.
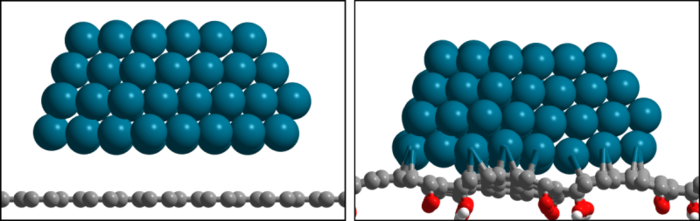
Abstract

16. M. Neurock*, Z. Tao, A. Chemburkar, D. Hibbitts, and E. Iglesia,
“Theoretical Insights into the Sites and Mechanisms for Base Catalyzed Esterification and Aldol Condensation Reactions over Cu.”
Faraday Discussions, 197 (2017) 59–86. Invited.
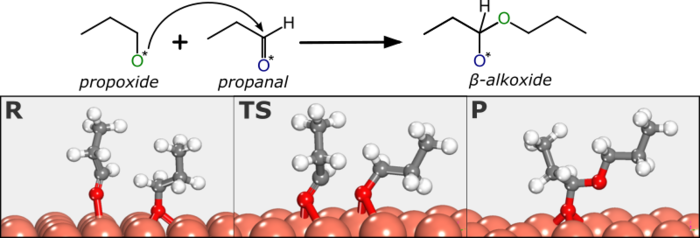
Abstract
2016
15. D. Hibbitts, D. Flaherty, and E. Iglesia*,
“Effects of Chain Length on the Mechanism and Rates of Metal-Catalyzed Hydrogenolysis of n-Alkanes.”
Journal of Physical Chemistry C, 120 (2016) 8125–8138.
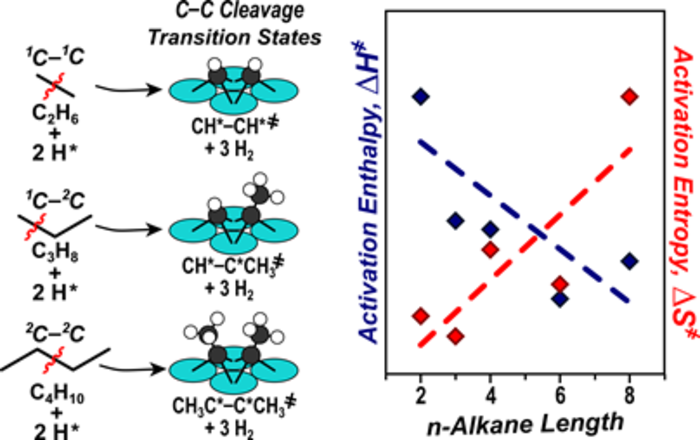
Abstract
14. D. Hibbitts, E. Dybeck, T. Lawlor, M. Neurock*, and E. Iglesia*,
“Preferential Activation of Carbon Monoxide near Hydrocarbon Chains during Fischer-Tropsch Synthesis.”
Journal of Catalysis, 337 (2016) 91–101.
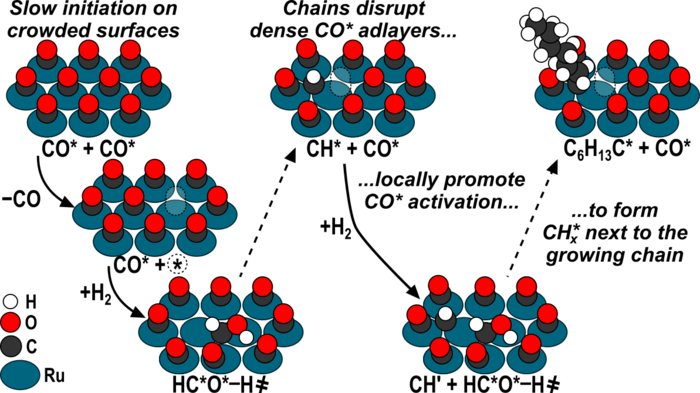
Abstract

13. D. Hibbitts and M. Neurock*,
“Promotional Effects of Chemisorbed Oxygen and Hydroxide in the Activation of C–H and O–H Bonds on Transition Metal Surfaces.”
Surface Science, 650 (2016) 210–220.

Abstract
12. D. Hibbitts, D. Flaherty, and E. Iglesia*,
“Role of Branching on the Rate and Mechanism of C–C Cleavage in Alkanes on Metal Surfaces.”
ACS Catalysis, 6 (2016) 469–482.
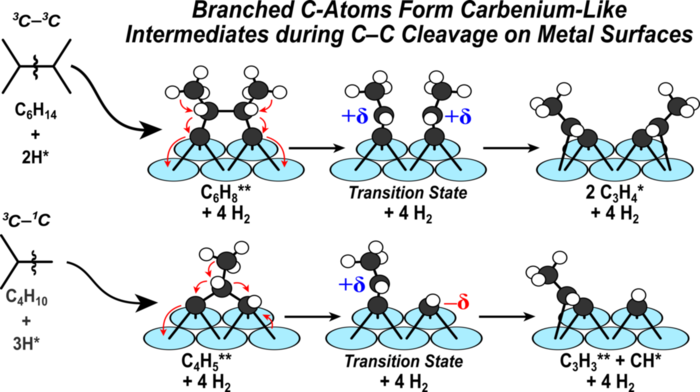
Abstract
2015
11. E. Gurbuz, D. Hibbitts, and E. Iglesia*,
“Kinetic and Mechanistic Assessment of Alkanol/Alkanal Decarbonylation and Deoxygenation Pathways on Metal Catalysts.”
Journal of the American Chemical Society, 137 (2015) 11984–11995.
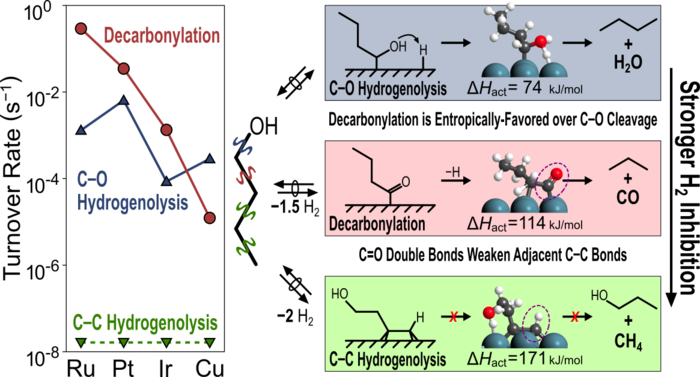
Abstract

10. D. Hibbitts and E. Iglesia*,
“The Prevalence of Bimolecular Routes in the Activation of Diatomic Molecules with Strong Chemical Bonds (O2, NO, CO, N2) on Catalytic Surfaces.”
Accounts of Chemical Research, 48 (2015) 1254–1262.
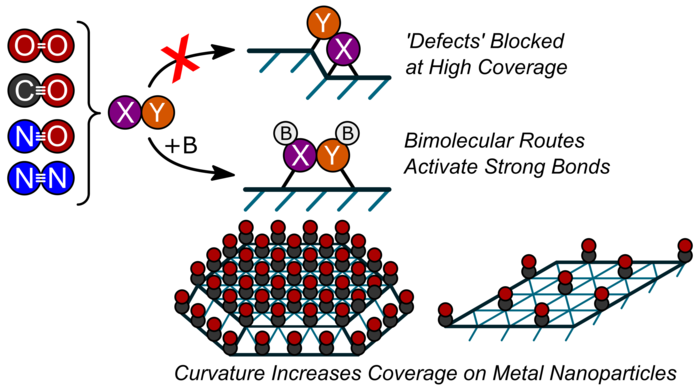
Abstract
2014
9. D. Hibbitts, R. Jimenez, M. Yoshimura, B. Weiss, and E. Iglesia*,
“Catalytic NO activation and NO‑H2 Reaction Pathways.”
Journal of Catalysis, 319 (2014) 95–109.
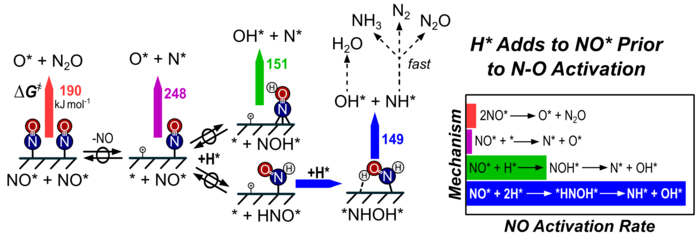
Abstract
8. D. Flaherty, D. Hibbitts, and E. Iglesia*,
“Metal-Catalyzed C–C Bond Cleavage in Alkanes: Effects of Methyl Substitution on Transition State Structures and Stability.”
Journal of the American Chemical Society, 136 (2014) 9664–9676.
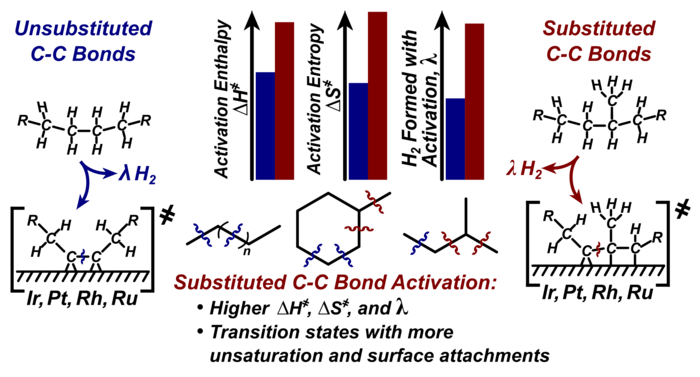
Abstract
7. D. Hibbitts, Q. Tan, and M. Neurock*,
“Acid Strength and Bifunctional Catalytic Behavior of Alloys Comprised of Noble Metals and Oxophilic Metal Promoters.”
Journal of Catalysis, 315 (2014) 48–58.
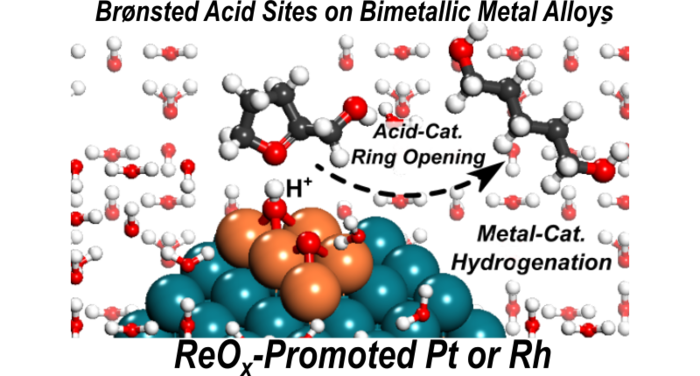
Abstract
6. D. Flaherty^, D. Hibbitts^, E. Gurbuz, and E. Iglesia*,
“Theoretical and Kinetic Assessment of the Mechanism of Ethane Hydrogenolysis on Metal Surfaces Saturated with Chemisorbed Hydrogen.”
Journal of Catalysis, 311 (2014) 350–356.
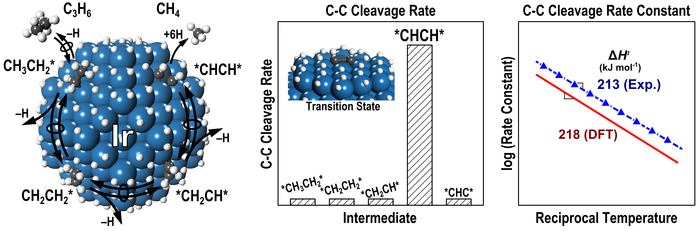
Abstract
2013
5. D. Hibbitts, B. Loveless, M. Neurock, and E. Iglesia*,
“Mechanistic Role of Water on the Rate and Selectivity of Fischer-Tropsch Synthesis on Ruthenium Catalysts.”
Angewandte Chemie, 52 (2013) 12273–12278.
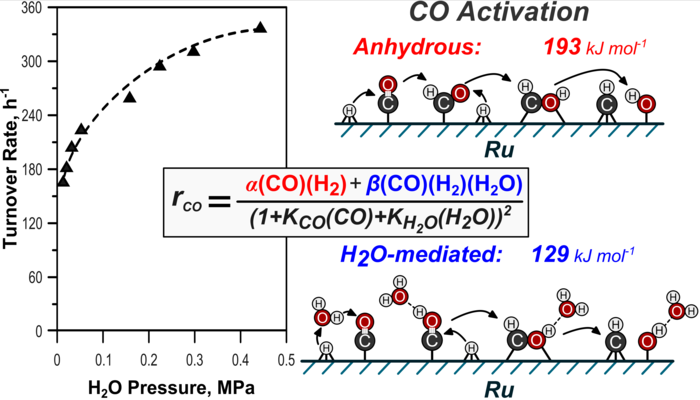
Abstract
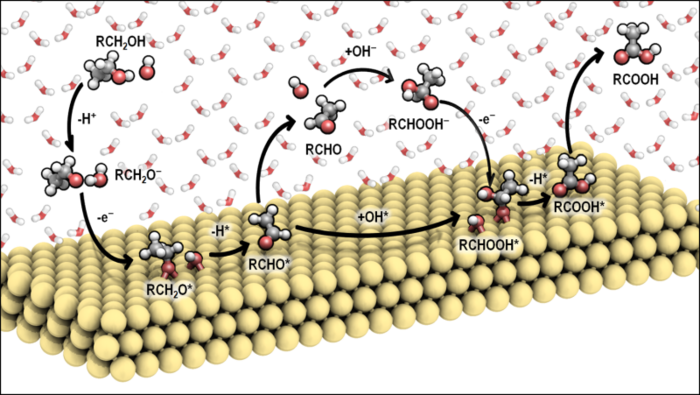
4. D. Hibbitts and M. Neurock*,
“Influence of Oxygen and pH on the Selective Oxidation of Ethanol on Pd catalysts.”
Journal of Catalysis, 299 (2013) 261–271.
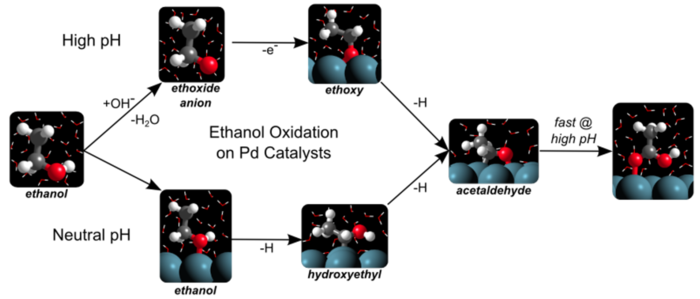
Abstract
2012

3. B. Braunchweig, D. Hibbitts, M. Neurock, and A. Wieckowski*,
“Electrocatalysis: a Fuel Cell and Surface Science Perspective.”
Catalysis Today, 202 (2013) 197–209.
Abstract
2011
2. M. Chia, Y. Pagan-Torres, D. Hibbitts, Q. Tan, H. Pham, A. Datye, M. Neurock, R. Davis, and J. Dumesic*,
“Selective Hydrogenolysis of Polyols and Cyclic Ethers over Bi-Functional Surface Sites on Rhodium-Rhenium Catalysts.”
Journal of the American Chemical Society, 133 (2011) 12675–12689.
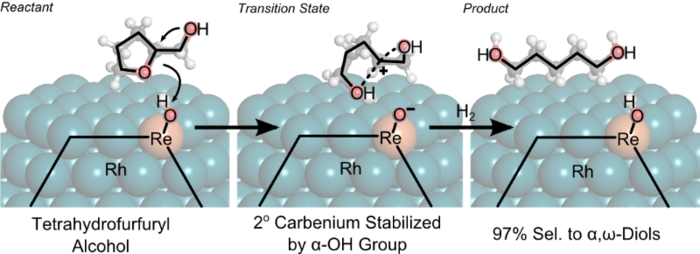
Abstract
2010

1. B. Zope, D. Hibbitts, M. Neurock, and R. Davis*,
“Reactivity of the Gold-Water Interface during Selective Oxidation Catalysis.”
Science, 330 (2010) 74–78.